
ISO 14971:2019 er en specialiseret standard omkring riskostyring indenfor medicinsk udstyr. På engelsk hedder den “Medical devices – application of risk management to medical devises.
I scope under punkt 1 angives det følgende på engesk:
“The process described in this document intends to assist manufactorers of medical devices to identify hazards associated with the medical device…”
Denne standard er altså en form for understandard til ISO 13485, men specifikt henvendt til producenter af medicinsk udstyr, da en meget stor del af standarden går på at producenten allerede under udvikling af det medicinske udstyr skal være opmærksom på de mulige farer der er ved brugen af udstyret, og skal forsøge at reducere og helst undgå sådanne farer, og farefulde situationer i at opstå.
Bemærk, at det som sagt ikke er et krav via ISO 13485, at en producent benytter denne standard, og det er heller ikke et krav via EU eller eksempelvis FDA i USA, at producenter følger denne standard. EU og USA har derimod defineret egne krav til risikostyring, og opfølgning via regulative krav. Det angives bl.a. i EU indenfor visse områder i regulativerne, at hvis producenten ikke benytter harmoniserede standarder (eksempelvis ISO standarder der er “accepteret” og offentliggjort som harmoniseret af EU), skal producenten som minimum leve op til de krav som disse standarder sætter indenfor det nævnte område. Se nærmere links til de europæiske regulativer nederst på denne side.
Bemærk ligeledes, at denne standard er en specifik standard til medicinsk udstyr, og at der findes en “generel” standard / vejledning til risikostyring der hedder ISO 31000:2018, som specificerer risikostyring på generelt plan.
STRUKTUREN:
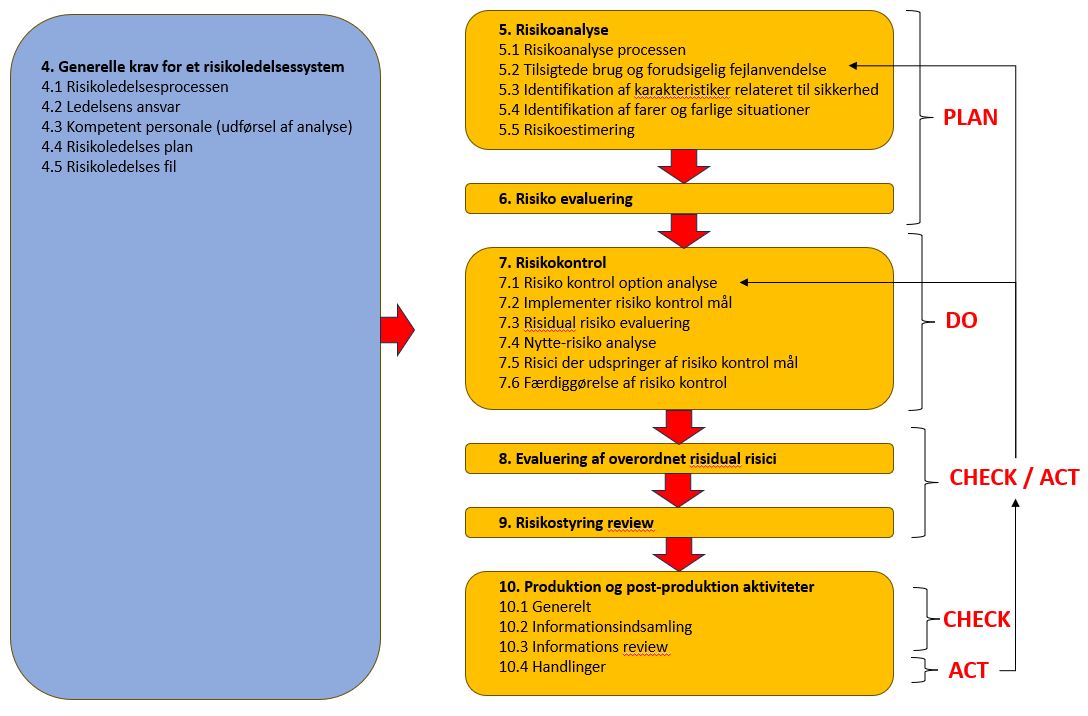
Klik på billede for at se dette
Punkt 4, angiver generelle krav til risikoledelsessystemet. Punkt 5 beskriver selve risikoanalysen, og punkt 6 beskriver risiko evalueringen, og punkt 7 risikokontrol.
Punkt 8 er evaluering af eventuel resterende risici, og punkt 9 er evaluering / review af den samlede risikostyring. Hvis ikke disse evalueringer medfører acceptable risici, skal man gå tilbage til eventuel punk 5.2 eller 7.1
Punkt 10 vedrører en overvågning når man er i produktion, og efter produktionen er gået i gang som kræver, at man overvåger markedet, og samler information om, og analyserer denne info, og agerer, hvis der kan identificeres problemer med det medicinske udstyr i markedet. Sidstnævnte krav omkring markedsovervågning er i tråd med kravene i EU regulativerne omkring medicinsk udstyr angivet i EU 2017/745 og EU 2017/746.